


 0
0

在铜催化的交叉偶联反应中,碳卤素键断裂的步骤仍然不明确,因为铜有多种氧化还原形态,而且形成的高价铜产物不稳定。

鉴于此,近期中国科学院上海有机化学研究所沈其龙、薛小松及加州大学伯克利分校John F. Hartwig共同通讯在Science 在线发表题为“Oxidative addition of an alkyl halide to form a stable Cu(III) product”的研究论文。
研究概述
该研究报道了α-卤代乙腈对离子和中性铜(I)配合物的氧化加成,形成以前难以捉摸但在这里完全表征了的铜(III)配合物。这些配合物的稳定性源于Cu−CF3键强和C(CF3)−C(CH2CN)键形成还原消除的高势垒。作者进行的机理研究表明,离子和中性铜(I)配合物的氧化加成通过两种不同的途径进行:离子配合物的SN2型取代和卤素原子转移到中性配合物。总之,该研究观察到明显的配体加速氧化加成,这与在铜催化偶联中观察到的唑、胺或炔与烷基亲电试剂的偶联有关。
图文导读
C -铜介导的交叉偶联反应已经成为构建碳-碳(C-C)和C-杂原子键的最有效方法之一。该领域早期的研究主要集中在sp2 -杂化碳亲电试剂的偶联上,近年来,研究范围扩大到sp3 -杂化碳亲电试剂的轻度偶联。近年来,在铜介导或催化的烷基卤化物的烷基化、芳化和胺化方面取得了很大进展,为在烷基链和环上安装官能团提供了一种替代和实用的方法。尽管这扩大了使用铜的烷基亲电试剂的交叉偶联反应的范围,但这些反应的机制尚不清楚。在此之前,提出了铜催化烷基亲电试剂交叉偶联反应的两个不同循环。一个循环包括一个双电子Cu(I)/Cu(III)歧管,一个循环包括一个逐步的Cu(I)/Cu(II)歧管,涉及Cu(I)中心和烷基亲电试剂之间的初始单电子转移(SET),生成Cu(II)中间体和烷基自由基,然后将官能团从生成的Cu(II)物种转移到烷基自由基。这两种途径的区分是具有挑战性的,因为反应中的高价铜中间体,特别是假定的Cu(III)中间体,具有高活性,通常无法检测。
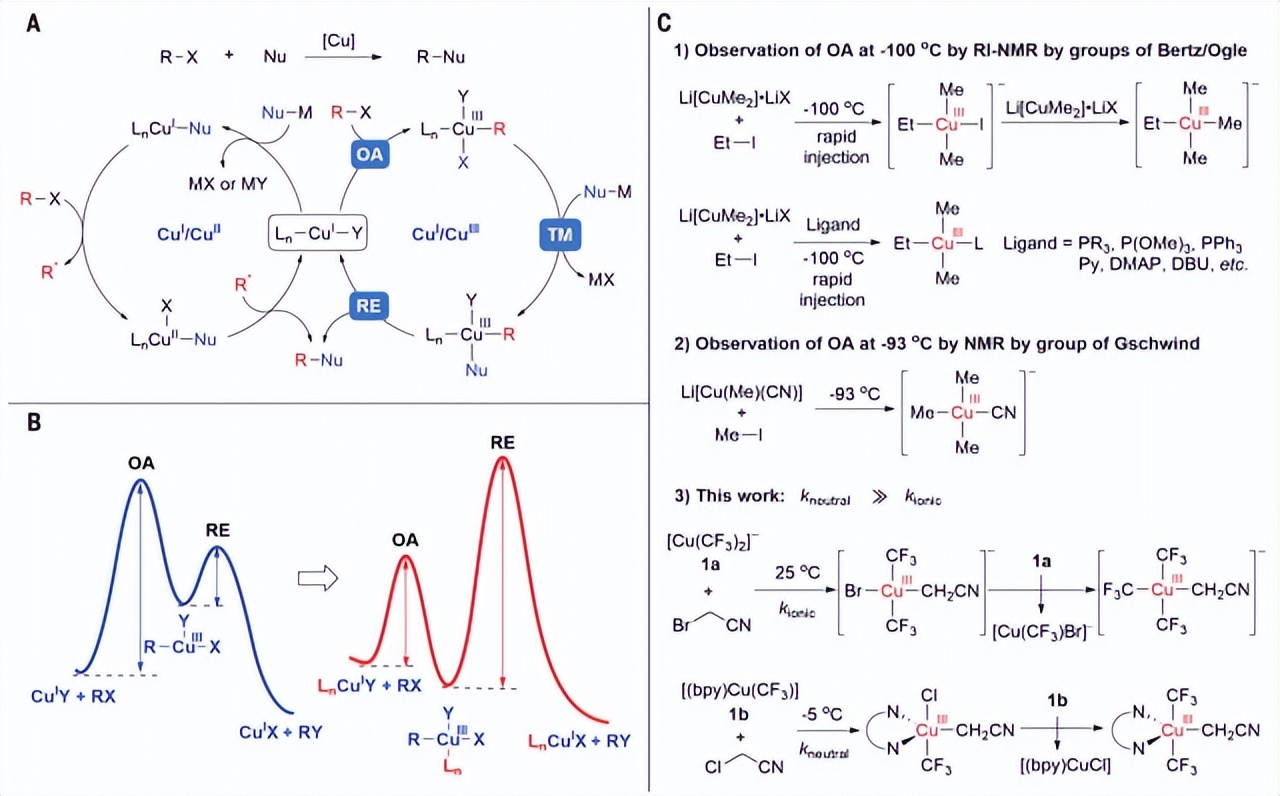
铜介导的交叉偶联机制(图源自Science )
因此,分离的,定义良好的烷基铜(III)配合物是罕见的。在21世纪初,Bertz、Ogle和Gschwind等人的研究表明,烷基碘化物与离子吉尔曼试剂反应会产生Cu(III)物质,这些物质在−93℃以下的温度下通过快速注入核磁共振(RI-NMR)光谱技术进行了表征。然而,即使在这个温度下,Cu(III)中间体的形成也很快,这使得烷基卤化物与Cu(I)的反应机理研究具有挑战性。
作者研究了烷基卤化物与三氟甲基-Cu(I)配合物的反应。最初选择稳定的[Ph4P]+ [Cu(CF3)2]-和[(bpy)Cu(CF3)] (bpy,联吡啶)作为Cu(I)配合物,分别代表无配体“ate”型Cu(I)配合物和中性的联吡啶化Cu(I)配合物。这些配合物将使作者能够探索两种不同类型的Cu(I)配合物的反应性差异以及氮配体对氧化加成过程的影响。作者选择了α-卤代乙腈XCH2CN[其中X(卤素)是Cl, Br或I]作为烷基亲电试剂,因为XCH2CN比其他烷基卤化物更亲电,降低了Cu(I)氧化加成的屏障。
由于氰基的吸电子性质,从[(配体)CuIII(CF3)2(CH2CN)]络合物中形成C−C键的还原消除势垒可以足够高,从而可以直接观察到产物,并可能分离出产物。该研究报道了从卤化乙腈氧化加成到离子和中性三氟甲基Cu(I)配合物中分离出Cu(III)配合物。对这些反应的机理研究,结合计算和实验研究,表明阴离子和中性配合物通过不同的机制与相同的烷基卤化物反应。
原文链接:https://www.science.org/doi/full/10.1126/science.adg9232
来源:化学科讯。声明:化浪化工网是出于传播信息之目的而转载此篇文章,若转载产生侵权,请作者持权属证明与本网站联系,我们将及时删除、更正,谢谢。您可关注化浪化工网获取更多化工相关资讯。如果您有对化工试剂、化学物质有采购需求,也可以登录化浪商城(自营商城)进行采购挑选。



















 浙公网安备
33020502000290号
浙公网安备
33020502000290号














